马金,丰宏林
(哈尔滨医科大学附属第一医院神经内科,哈尔滨 150001)
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)是一种选择性累及上、下运动神经元的致命性神经系统变性疾病[1],主要累及锥体束、脑干和脊髓前角细胞,临床表现为进行性加重的肌肉萎缩、无力、痉挛以及认知损害等,通常中年以后发病,表现为持续性、进行性肌肉萎缩和无力,多数患者因呼吸机麻痹在发病后2~4年死亡[2]。全球ALS的发病率逐渐升高,该病不仅给患者带来巨大痛苦,而且给患者家庭及社会造成了沉重负担。由于ALS致病机制尚未明确,因此有效治疗措施的研究也进展缓慢。研究显示,基因突变参与ALS的发病,并与疾病进展密切相关[3]。但很多导致或使人易患ALS的基因及其变异仍然未知,因而尚不能完全明确ALS的具体发病机制,这也给疾病的治疗和新药研发带来较大挑战。现就与ALS发病及进展相关的基因及其潜在的分子遗传学机制进行综述,拟为ALS更深入的致病机制研究及致病基因发现提供参考,亦为相关新药的作用靶点及诊断标志物的开发与利用提供依据。
ALS是运动神经元退变性疾病,其中10%为常染色体显性遗传,呈家族性分布,其余约90%是散发性ALS,家族性ALS的部分遗传基因也存在于散发 ALS中[3-4]。随着分子遗传技术在ALS研究中的应用,发现40%~55%的ALS由已知的ALS相关基因变异引起,目前已发现的与ALS发病相关的基因约有50个[3,5],见表1,其中最常见的为超氧化物歧化酶1(superoxide dismutase 1,SOD1)突变(20%)、C9orf72(chromosome 9 open reading frame 72)重复扩增(40%)、肉瘤融合蛋白(fused in sarcoma,FUS)(1%~5%)和交互反应DNA结合蛋白-43(transactive response DNA binding protein-43,TDP-43)(1%~5%)[6-7]。人种不同,致病基因的变异亦不同,其中欧洲人以C9orf72突变为主,占33.7%,其余依次为SOD1(14.8%)、TDP-43(4.2%)、FUS(2.8%);
亚洲人以SOD1突变为主(30%),其余依次为FUS(6.4%)、C9orf72(2.3%)、TDP-43(1.5%)[5]。中国人群中ALS相关致病基因总体突变率在家族性ALS中为55.0%,在散发性ALS中为11.7%,其中SOD1基因突变频率最高(25.6%),其次为FUS(5.8%)、TDP-43(5.8%)、DCTN1(Dynactin 1)(3.6%)以及C9orf72(3.5%)[8]。ALS的发病可能与氧化应激、线粒体功能障碍、兴奋性氨基酸毒性、内质网应激、轴突运输障碍、RNA代谢紊乱、神经炎症及胶质细胞病变等有关,这些致病机制与ALS相关致病基因变异密切相关[3]。
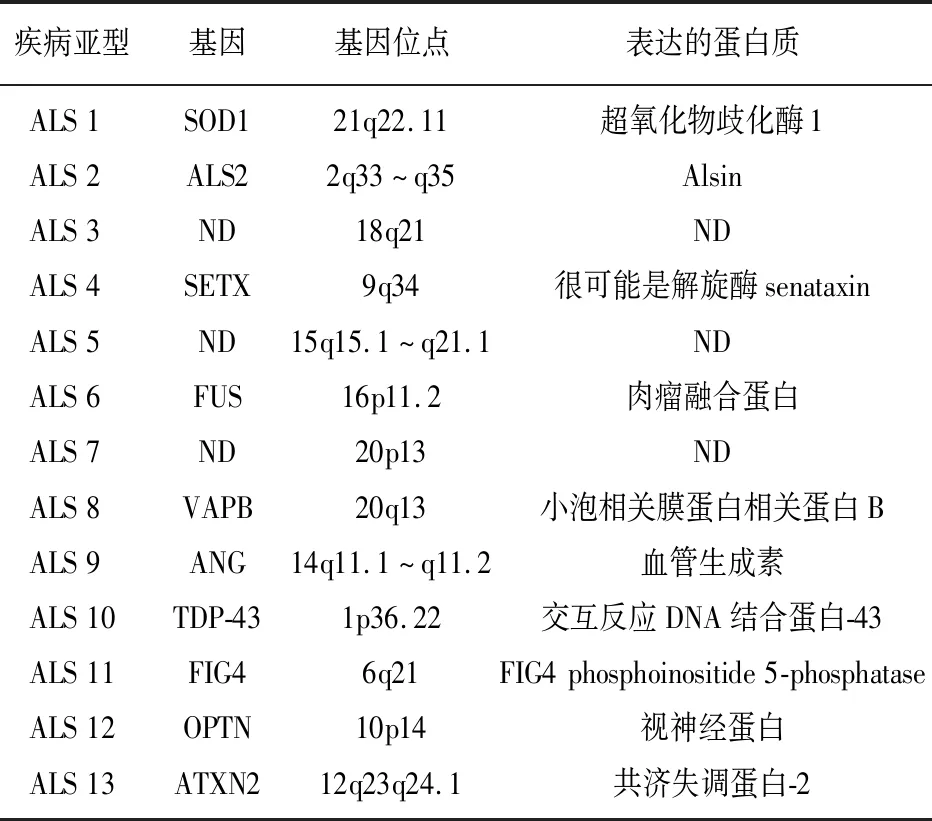
表1 与ASL发病相关的基因、基因位点以及表达的蛋白质
2.1SOD1 在ALS相关致病基因中,SOD1是报道最多且研究最为深入的基因。SOD1基因与ALS具有关联性,其编码一种含有153个氨基酸的金属酶[9]。SOD1二聚体存在于细胞质、细胞外部空间及线粒体外膜,通过将细胞呼吸过程中产生的超氧阴离子自由基歧化为O2和H2O2,而起到保护细胞免受活性氧损害,保持细胞内活性氧化物内稳态的作用[2]。目前研究已报道超过180种与ALS相关的SOD1基因变异,这些基因变异与疾病病程、表型及严重程度相关[10],如发生A4V、H43R、L84V、G85R N86S和G93A变异者疾病进展迅速,患者生存时间短;
而发生G93C、D90A或H46R变异者通常寿命较长[11]。SOD1基因特定变异者在ALS中表现出不同的临床特征,如A4V变异与四肢发作的侵袭性ALS相关,D90A纯合子患者通常表现为缓慢进展的轻瘫,而杂合子D90A变异与延髓、上肢和下肢等运动相关区域功能障碍相关,且进展较快[12]。
SOD1基因突变致使SOD1的构象和功能发生改变,从而导致ALS。研究指出,SOD1基因变异与多种蛋白质的作用相关,包括兴奋性毒性、活性氧上调引起的氧化应激、内质网应激、线粒体功能障碍等[13]。SOD1基因突变体改变机体的氧化活性,可导致有毒的羟自由基累积,破坏蛋白质折叠并损害运动神经元的轴突运输[14]。此外,SOD1基因突变体可诱导内质网应激,导致未折叠蛋白反应和内质网相关降解激活,内质网的相关降解可破坏细胞稳态,促进细胞凋亡,引起运动神经元的损伤[7]。SOD1突变可引起轴突运输损害,错误折叠的SOD1不能通过线粒体膜运输,而是在线粒体外膜积累,从而触发线粒体依赖的细胞凋亡程序,引起运动神经元凋亡[15]。亦有研究显示,野生型SOD1蛋白的错误折叠是导致ALS病情进展的重要因素[16]。
目前对于SOD1变异产生兴奋毒性的形式尚存在争议,争议主要集中在SOD1蛋白是可溶性形式还是聚集性形式。可溶性SOD1蛋白可在细胞核及细胞质中穿梭,随着可溶性SOD1蛋白累积的增加,细胞对抗应激刺激的生理防御增加,从而抑制超氧阴离子增加引起的DNA损伤[17]。研究显示,细胞核中可溶性SOD1水平越高,ALS的病程持续时间越长,两者呈正相关,表明可溶性SOD1蛋白在核室中可能具有运动神经元保护作用[17]。也有学者发现,突变或外界因素导致SOD1聚集性形式增加可导致细胞毒性增加,聚集性SOD1累积产生的毒性可破坏细胞通路,导致线粒体功能障碍、兴奋性毒性、氧化应激、内质网应激、轴突运输中断和细胞间朊病毒样增殖[10,13-17],提示累积产生的毒性可能在ALS的发病中起重要作用。以上发现强调以减少聚集性SOD1为目标治疗ALS的潜在好处。
2.2TARTDP-TDP-43 TARTDP基因编码TDP-43[18],其是人体内广泛存在的一类DNA转录抑制因子。TARTDP在脊髓运动神经元中的主要功能是通过与低分子量微丝信使RNA(message RNA,mRNA)结合,进而影响微丝与细胞支架的形成[18]。TDP-43是由414个氨基酸组成的DNA/RNA结合蛋白,通常在细胞核与细胞质之间穿梭,是导致ALS患者蛋白聚集的主要因素[19]。迄今为止,在不同种族中发现了40多个TARDBP变异体,大部分变异是位于转录本C端富含甘氨酸区域的错义变异[18]。研究显示,在无TARDBP基因变异的散发病例和C9ORF72变异者的脑和脊髓中泛素阳性包涵体中存在TDP-43的聚集[19-20]。TDP-43聚集成团,导致RNA代谢的正常功能丧失,进而导致脑细胞死亡或脊髓细胞神经毒性[21],表明TDP-43基因变异是导致ALS和神经退行性变的主要原因。
TDP-43的致病机制比较复杂。TDP-43作为基因表达的调节因子参与RNA加工,包括前mRNA剪接、mRNA稳定性调控、mRNA转运、翻译以及非编码RNA调控[22-23]。TDP-43与前mRNA的3′非翻译区结合,通过反馈机制自我调节:当TDP-43过剩时,mRNA转录产物被降解而不是被翻译,致使细胞功能障碍;
当TDP-43减少或耗尽时,mRNA代谢广泛失调[19]。TDP-43在转录翻译中的乙酰化、泛素化、磷酸化、蛋白水解及二硫键形成等亦被认为是ALS的潜在致病因素[24-25]。再者,细胞质中TDP-43上调形成的包涵体在细胞间传播,进而损伤神经元,导致ALS患者的运动神经元死亡和神经退行性病变[26-27]。此外,有研究报道细胞核中TDP-43的减少会抑制运动神经元轴突的再生,运动神经元中TARDBP/TDP-43的上调通过改变RNA的剪接和稳定性增加ALS的发生风险;
同时TARDBP/TDP-43缺失通过下调骨骼肌中TBC1D1(TBC1 domain family member 1)蛋白导致年龄依赖性脑萎缩,进而导致神经元功能受损[7,27]。
2.3FUS FUS基因编码由526个氨基酸组成的多功能蛋白,属于家族性特发性震颤RNA结合蛋白,2009年发现FUS基因与ALS相关,主要参与基因转录和表达的调控[28-29]。FUS基因突变多呈常染色体显性遗传,与早发型和青少年ALS有关[30-31]。目前发现的与ALS相关的FUS突变超过50种[2]。在正常生理条件下,FUS主要定位于细胞核,穿过细胞质参与核质的运输,通过前mRNA剪接、RNA转运和翻译在基因表达中发挥作用,FUS亦参与DNA修复,包括DNA双链断裂修复中的同源重组和非同源端连接,并在细胞的副斑点形成中发挥防御作用[23,32-33]。因此,突变的FUS会破坏核质运输,致使细胞核耗尽,而FUS在细胞质中聚集可导致神经毒性[34]。FUS突变不仅抑制了蛋白质的整体翻译,也抑制了蛋白质的局部翻译,使树突和轴突的终末受损;
同时FUS突变也会导致突触稳态缺陷和功能障碍,突触稳态维持所需蛋白的减少也可能导致ALS[35]。
运动神经退行性病变与细胞中可溶性FUS聚集直接引起的毒性相关[36]。家族性ALS中有约5%的患者会出现融合在FUS中的RNA结合蛋白突变,在这些患者中可观察到FUS阳性细胞质包涵体[33]。进一步的动物实验表明,体内FUS与RNA的相互作用可增加FUS在神经元细胞质中的聚集,加重疾病的严重程度[37]。此外,FUS变异是导致细胞功能受损而致病的直接因素,其会导致RNA剪接缺陷、DNA破坏及FUS的不能自动调节[33,38-39]。研究亦表明,FUS基因突变导致的病理性折叠蛋白可引发朊毒体的形成,导致神经退行性病变,如ALS[40]。需要特别关注的是,FUS相关ALS的致病机制是细胞核内FUS生理功能的丧失,而不是细胞质FUS聚集毒性的丧失[7]。
2.4C9orf72 C9orf72首先在欧洲人群中发现,是基因非编码区六核酸[(GGGGCC)n]重复扩增序列,其在亚洲人群中并不常见[2]。研究显示,C9orf72在ALS患者中反复扩增,mRNA和蛋白水平下降,进而导致与疾病相关的蛋白缺失[41-42]。研究显示,重复序列的G碱基增加会使转录中易形成G四倍体二级结构,从而引发一系列蛋白质的异常相互作用[41]。对ALS中与C9orf72相关通路的研究显示,RNA的错误处理是关键。首先,改变了对扩增的转录碱基本身的处理:C9orf72转录碱基包含两个交替使用的第一外显子(1a和1b),重复扩增存在于它们之间的内含子中。转录产物产生的各种异构体包含一个或两个外显子,其存在有利于1a外显子转录的重复扩增。这导致含有重复扩增的转录产物比例增加,影响内含子重复转录的RNA处理,如转录失败、内含子重复剪接减少和核聚集[43-44]。其次,部分含有重复转录的碱基受到非血管紧张素原的RNA翻译,导致异常的双肽重复蛋白产生,进而在中枢神经系统形成神经包涵体,并可能参与疾病的发生[45]。再次,扩增的重复序列会对RNA加工产生影响。大部分C9orf72-ALS患者中发现了含有重复扩增的RNA,这些RNA位点能够与各种RNA结合蛋白发生异常相互作用,进而对RNA的表达和剪接产生影响,从而产生毒性[20,46]。
C9orf72的致病机制复杂多样,除上述蛋白丢失理论及对RNA的影响外,C9orf72还可通过诱导运动神经元中甘氨酸-精氨酸重复蛋白表达,增加DNA损伤、氧化应激及线粒体功能障碍[47];
C9orf72突变可影响泛素-蛋白酶体系统,进而诱导运动神经元死亡[48];
C9orf72变异可通过激活α-氨基-3-羟基-5-甲基-4-异唑丙酸受体,致使运动神经元通过影响神经胶质细胞对谷氨酸的摄取,使谷氨酸兴奋性增加,从而导致运动神经元的损伤甚至死亡[49]。
2.5其他基因变异 随着基因测序技术的发展,大量与ALS相关的基因被发现,这些变异主要影响RNA稳态与转运、蛋白质稳态和细胞骨架动力学。这些基因变异虽然罕见,但也为ALS的致病机制研究提供了一些线索,其中与RNA加工相关的基因有血管生成素、SETX、基质蛋白3、共济失调基因2型、TAF-15、EWSR1、异质核核糖核蛋白A1、不均一核糖核蛋白、延长复合体蛋白3,与蛋白质运输和降解相关的基因有鸟嘌呤核苷酸交换因子2、囊泡相关膜蛋白相关蛋白B/C、CHMP2B、FIG4、UBQLN2、SQSTM1、SIGMAR1、视神经蛋白、缬络胺酸蛋白质、TANK结合激酶1,与细胞骨架和轴突动力学相关的基因有DCTN1、前纤维蛋白1、SPG11、TUBA4A、NEFH、外周蛋白、NEK1,与线粒体相关的基因CHCHD10等[7]。基因变异影响ALS的表型及易感性。Diekstra等[50]研究显示,UNC13A基因突变与ALS的易感性及患者生存期相关。共济失调基因1和共济失调基因2的三核苷酸重复扩增会增加患病风险[51-52]。Uyan等[53]研究显示,EPHA3基因的变异表达与ALS发生呈负相关,被认为是ALS的潜在保护因素。综上,ALS的发病与基因变异或遗传背景有密切关系。
最近发现了一系列与ALS相关的基因。DNAJC7编码热激蛋白70,其在蛋白折叠、稳定等功能中发挥关键作用,可导致ALS模型中蛋白聚集[54]。WDR7的最后一个内含子可能与ALS有关,其扩增可导致RNA聚集[55]。酰基辅酶A合成酶长链家族成员5是一个作用于神经A1星形胶质细胞的基因,可诱导A1星形胶质细胞产生神经毒性,导致运动神经元死亡和ALS进展,酰基辅酶A合成酶长链家族成员5在ALS中的表达上调,其过表达与人类体重快速减轻有关[56]。Sigma-1受体可调节线粒体呼吸,控制细胞对内质网和氧化应激的防御,Sigma-1受体的过表达具有神经毒性,导致线粒体功能障碍,在ALS致病中发挥作用[57]。GLT8D1也被认为是ALS的致病基因,其是糖基转移酶8家族的一员,参与催化糖基的转移。GLT8D1突变可抑制糖基转移酶的正常活性,并对神经节苷脂信号转导产生负面影响,该基因的变异与ALS患者的疾病严重程度和细胞毒性呈正相关[58]。KIF5A是一个与ALS相关的新基因,KIF5A突变可影响ALS患者的发病年龄、存活时间,KIF5A的变异破坏了轴突运输,导致突触中淀粉样前体蛋白缺失,从而导致神经退行性病变[59]。虽然上述基因已被证实是ALS致病基因,但研究仍然有限,尚不完全清楚这些基因在ASL中的详细作用机制。
自发现SOD1突变与ALS致病相关以来,ALS致病的分子遗传学机制已经取得一系列进展。但由于ALS的病因病机十分复杂,除遗传外,与生活习惯(活动及劳动强度等)、环境(紫外线、特殊物质接触史)等也密切相关,因此目前难以对ALS的发病机制做出全面的诠释。对新的致病基因、基因修饰及其分子途径的研究可能会提高人们对ALS的认识。此外,对ALS致病基因间潜在的相互作用进行研究有助于其靶向治疗。目前针对ALS提出了为单个患者提供个性化反义寡核苷酸治疗的治疗理念,在此基础上,为ALS的常见致病途径寻找靶点药物已成为未来研究的重要方向,而个体化多靶点的干预治疗较单靶点标准化治疗可能更使患者受益。
猜你喜欢 运动神经元线粒体毒性 线粒体质量控制在缺血性脑卒中的作用研究进展医学研究生学报(2022年5期)2022-12-07“60%敌畏马乳油”农药对家蚕残毒性调查四川蚕业(2022年2期)2022-11-19线粒体自噬在纤维化疾病中作用的研究进展中华实用诊断与治疗杂志(2022年1期)2022-08-31除草剂敌草快对克氏原螯虾(Procambarus Clarkii)的毒性研究当代水产(2022年6期)2022-06-29运动神经元的模型研究方法体育科技文献通报(2021年12期)2021-12-18线粒体自噬在蛛网膜下腔出血中的研究进展中国卒中杂志(2021年7期)2021-11-29应激宁小鼠急性毒性试验及亚慢性毒性试验当代水产(2021年6期)2021-08-13甲基苯丙胺神经毒性作用及机制的研究进展昆明医科大学学报(2021年2期)2021-03-29A Miracle of Love考试与评价·高二版(2020年2期)2020-09-10为什么负重后手臂会发抖?科学之谜(2019年3期)2019-03-28